
Проблема діагностики спадкових дисплазій сполучної тканини, зокрема синдрому Марфана (СМ), залишається актуальною. Часто встановити правильний клінічний діагноз є складно, пацієнт вже в дорослому віці взнає про свою проблему, що в кінцевому результаті призводить до важкості корекції соматичних змін. Це з одного боку зумовлене важкістю діагностики, з іншого – неточним або неправильним трактуванням результатів додаткових методів обстеження.
Синдром Марфана – захворювання з аутосомно-домінантним типом успадкування, яке відноситься до групи спадкових фібрилінопатій, спадкової патології сполучної тканини зі змінами скелету, органа зору та серцево-судинної системи.
В основі СМ є дефект гена фібриліну-1 (FBN1), який кодує білок позаклітинного матриксу, що входить до складу еластичних волокон.
Ще у 1896 році французький педіатр Антуан Жан Марфан описав п’ятирічну дівчинку із надзвичайно тонкими і довгими кінцівками, контрактурами суглобів, кіфосколіозом і назвав цей синдром pattes d’araignee (пальці павука).
Основні прояви СМ:
- Ураження скелету і лицевого черепа (деформація грудної клітки або асиметрія грудей, довгі пальці, високий зріст, сколіоз або кіфоз, готичне піднебіння, криві зуби).
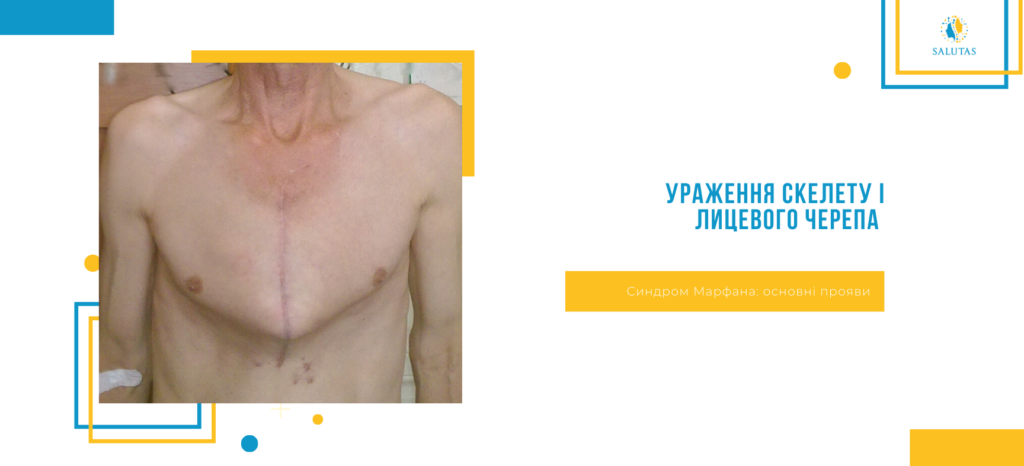
- Розтяжки на шкірі (шкірні стрії), особливо на спині.
- Зміни серцево-судинної системи – пролапс мітрального клапана, двостулковий аортальний клапан, збільшення розміру аорти на рівні синусів Вальсальви.
- Зміни очей – підвивих кришталика, міопія, плоска рогівка, астигматизм.
Дуже важливо при огляді пацієнта з СМ проводити системну оцінку стану хворого на СМ, так звану (Systemic score). Вона є доступна онлайн і кожен лікар може її порахувати.
Важливим методом діагностики є ехокардіографія. Кожен пацієнт з симптомами подібними на СМ має звернутися до кардіолога для оцінки його стану і пройти ехокардіографію на предмет ураження аорти і клапанів серця. Наступною має бути консультація офтальмолога.
Основним методом діагностики СМ є генетичне дослідження. Пацієнтам після проведених раніше кардіохірургічних втручань на аорті, а також пацієнтам з клінічними ознаками СМ або іншими специфічними дисплазіями сполучної тканини для підтвердження діагнозу СМ потрібно провести альтернативне генетичне тестування на визначення 29 генів, що беруть участь у формуванні аневризм грудної аорти.
Така послуга доступна сьогодні в Україні. У МЦ Салютас Ви можете здати аналіз на генетичне тестування для діагностики спадкових дисплазій сполучної тканини. Саме секвенування проводиться у відомій американській лабораторії.
У лікування СМ використовують медикаментозне і хірургічне лікування. Медикаментозна терапія повинна бути продовжена і після операції.
Серед медикаментів застосовують β-блокатори, які знижують швидкість
розширення аорти. Метою лікування має бути суворий контроль за артеріальним тиском (систолічний тиск до 120 мм рт. ст. і 110 мм рт. ст. для пацієнтів з розшаруванням аорти). Більшість центрів світу використовують бісопролол.
Також використовують блокатори рецепторів ангіотензину II, зокрема лозартан, які призводять до TGF-β антагонізму і профілактики аневризми аорти у хворих на СМ.
Для ефективного диспансерного обліку та ведення хворих на СМ у МЦ Салютас впроваджено в клінічну практику покроковий алгоритм, який передбачає стандартизований підхід до ведення хворих на СМ.
- • 1-й крок – постановка діагнозу СМ;
- •2-й – оцінка якості життя і необхідність проведення додаткових обстежень;
- •3-й – зміна способу життя і обмеження різких фізичних навантажень;
- •4-й – прийом профілактичних медикаментозних засобів (бісопролол 5 мг або лозартан 100 мг на добу) для контролю дилятації аорти;
- •5-й – планове оперативне втручання на аорті у разі прогресування дилятації або ознак розшарування;
- •6-й щорічне динамічне спостереження з консультацією кардіолога, кардіохірурга, офтальмолога.
Профілактика ускладнень у хворих на СМ найефективніший спосіб боротьби з хворобою. Не зволікайте, Звертайтеся, завжди раді Вам допомогти.